
GeneLinea's goal is to be very easy to use, so you as a biologist can spend more time focusing on your data rather than learning the command line interface and programming. But we all run into problems now and again. This is the help/FAQ section that can hopefully guide you. If you continue to have issues please email the author.
If you are new to GeneLinea and want to see how it works and do not currently have a dataset to work with, here are some files you can play with to see the framework in action. View more to get the download link:
The file format input must be Genbank (.gbk or .gb) format post annotation (automatic or manual). We have used Prokka for in-house use on our bacteria, and runs flawlessly for rapid annotation of prokaryotic genomes in GeneLinea. Here is a list of possible other annotation pipelines. Although we use the FASTA sequence from the Genbank to run alignments - we use the Genbank annotations for the gene names. Your mileage may vary on results because of the irregularities. It is best to stay consistent (same annotation pipeline on all genomes). Often when GeneLinea fails, it is because of a poor annotation. To see a list of annotation pipelines, click details:
GeneLinea run best when using finished sequence - it becomes less reliable with draft sequences, or poorly assembled genomes. Following the rule: Garbage in garbage out. Think about that in life too. To read more about finished sequence click on details:
So how long will it take to run my stuff? Well, that depends on a lot of things. But we really tried to get the programming efficient as possible to prevent wait a long time. Also something to keep in mind, we programmed it with bacterial genomes in mind (~5MB) - anything more than this may take some serious time. But if your working with large genomes, you are probably already used to this.
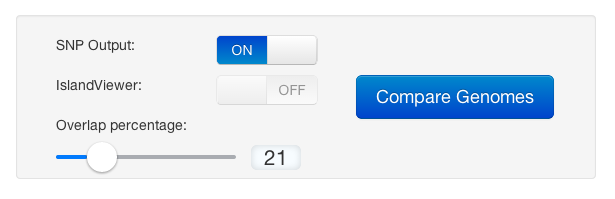
We were trying to keep many options automated so this would be straightforward process, and good for iterations (adding/subtracting genomes, etc). This is what we have boiled the user options down to: